The increasing demand for complex functional molecules in pharmaceuticals, advanced materials, and chemical technologies places growing pressure on the development of synthetic methods that are efficient, selective, and sustainable. Many of the transformations that underpin modern molecular manufacturing still rely on energetically demanding conditions or narrow reactivity paradigms, which limit flexibility, scalability and long-term sustainability. In this context, the ability to control chemical reactivity using light and catalytic systems offers a powerful opportunity to redesign how molecular complexity is generated.
Our research program aims to develop light-driven and catalytic platforms that enable precise control over bond formation, selectivity and reaction pathways. By integrating photoredox catalysis, transition-metal catalysis and computational modeling, we seek to uncover unconventional reactivity modes and translate them into robust and broadly applicable synthetic methodologies. Visible light is employed as a programmable and traceless energy input to access reactive intermediates and mechanistic manifolds that are difficult or inaccessible under classical thermal conditions, thereby expanding accessible chemical space under mild and operationally simple conditions.
Within this platform, we explore visible-light-driven radical processes for selective functionalization and molecular assembly, as well as the photochemical and catalytic activation of azobenzenes as programmable molecular building blocks. These studies demonstrate how light can be leveraged not only as a trigger for bond formation but also as a tool to control reaction pathways and molecular outcomes. Overall, our research lies at the interface of organic synthesis, catalysis, photochemistry, and computational chemistry, and seeks to deliver conceptually innovative and practically useful solutions relevant to academic, industrial, and societal challenges.
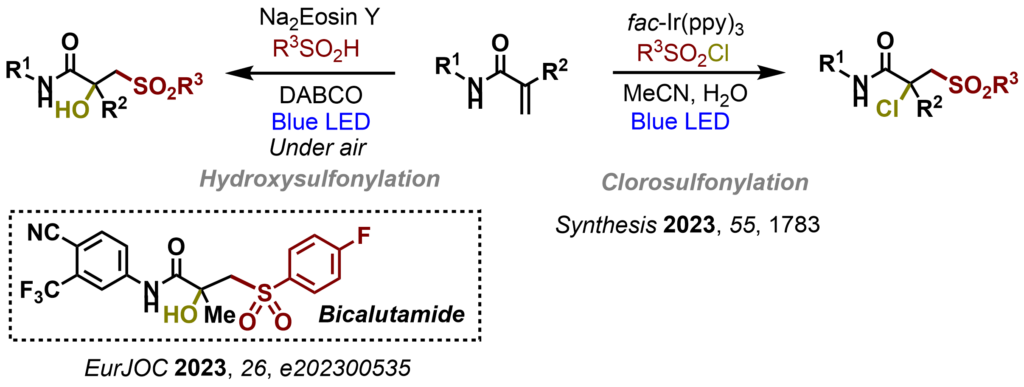
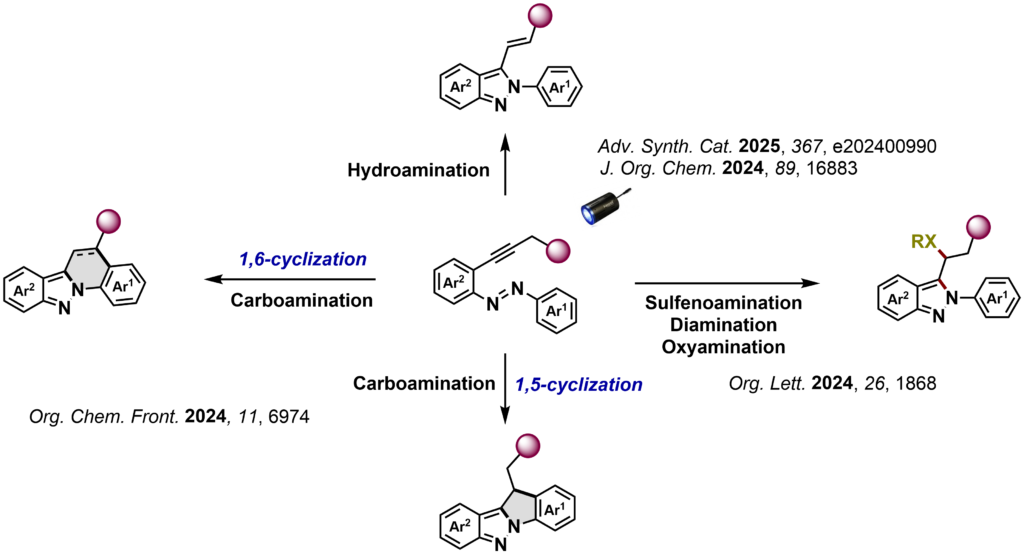
A central element of our strategy is the tight coupling of experiment and theory. Density functional theory (DFT) calculations are used to rationalize reactivity trends, identify selectivity-determining steps and guide catalyst and reaction design, enabling a predictive approach to reaction development (For example: ChemCatChem 2024, 16, e202400909; Angew. Chem. Int. Ed. 2024,63, e202319158; Nat. Chem. 2024, 16, 607-614; J. Org. Chem. 2023, 88, 14131; ACS Macro Lett. 2023, 12,949; ChemSusChem, 2023, 16, e202300200; Org. Biomol. Chem. 2023, 21, 2705; Angew. Chem. Int. Ed. 2022, 61, e202205651; Chem. Sci.2021, 12, 15084-15089).